Progeria es una enfermedad
genética de la infancia extremadamente rara, caracterizada por un envejecimiento brusco y prematuro en niños entre
su primer y segundo año de vida. Se estima que afecta a uno de cada 8
millones de recién nacidos vivos. No se ha evidenciado preferencia por
ningun sexo en particular, pero se han comunicado muchos mas pacientes
de raza blanca. La progeria puede
afectar diferentes órganos y tejidos: hueso, músculos, piel, tejido
subcutáneo y vasos.
La forma más severa de esta enfermedad es la llamada síndrome de
Hutchinson-Gilford.
Características clínicas
Baja estatura
Piel seca y arrugada
Calvicie prematura
Canas en la infancia
Ojos prominentes
Cráneo de gran tamaño
Venas craneales sobresalientes
Ausencia de cejas
y pestañas
Nariz grande y con forma de pico
Mentón retraído
Pecho angosto, con costillas marcadas
Extremidades finas y esqueléticas
Estrechamiento de las arterias coronarias
Articulaciones grandes y rígidas
Manchas en la piel semejantes a las de la vejez por mal metabolismo
de la melanina
Presencia de enfermedades degenerativas como la artritis
o cataratas,
propias de la vejez
Muerte natural hacia los 13 años.
Mitosis con retardo reticoendoplasmatico.
Falta de gametos sexuales y pensativos.
Alteración en la dentición
Osteólisis del tercio distal de las claviculas
Osteólisis de falanges distales en manos y pies
Osteoporosis
Arterosclerosis
Pronóstico
El promedio de vida en niños enfermos es de 13 años, pero puede estar
entre 7-27 años, aunque la supervivencia más allá de la adolescencia es
inusual, se ha descrito un paciente que falleció a los 47 años por un
infarto del miocardio. En más del 80% de los casos la muerte
se debe a complicaciones que surgen, como la arteroesclerosis,
fallos en el corazón, infarto del miocardioy trombosiscoronaria.
Tratamiento
No existe aún un tratamiento de probada eficacia. La mayoría de los
tratamientos se limitan a paliativos o prevención de complicaciones como
son las enfermedades cardiovasculares. Se utilizan aspirinas en bajas
dosis y dietas hipercalóricas. Se han intentado tratamientos con hormona
de crecimiento humano.
Después de ser descubierto el gen causante de ella y su mecanismo se
ha propuesto un tratamiento con un tipo de droga anticancerígena,
inhibidora de la farnesyltransferasa (FTIs), se ha probado su eficacia
en modelos con ratones.
A partir de Mayo de 2007 se inició un período de pruebas clínicas con
pacientes utilizando FTI Lonafarnib. Aunque recientemente se ha descubierto específicamente el gen
causante de la progeria, aún no existe cura. El factor de que los
pacientes mueren jóvenes, casi siempre en la segunda década de vida, no
ayuda en el descubrimiento de la cura, ya que no se pueden llevar a cabo
estudios más completos y especializados, los cuales tardarían años.
El síndrome de Rett es una enfermedad congénita con compromiso neurológico
que afecta la gran mayoría de las veces a sujetos del sexo femenino.
La enfermedad no es evidente en el momento del nacimiento, se
manifiesta generalmente durante el segundo año de vida, y en todos los
casos antes de los 4 años. Afecta aproximadamente a 1 niña de cada
10.000 nacidas vivas. Puede observarse retraso grave en la adquisición
del lenguaje
y de la coordinación motriz, así como retraso mental grave o severo. La
pérdida de las capacidades es por lo general persistente y progresiva.
El síndrome de Rett provoca grave discapacidad
en todos los niveles, haciendo al enfermo dependiente de los demás para
el resto de la vida.
Síntomas
Tras una fase inicial de desarrollo normal, se asiste a una detención
del desarrollo y luego a un retroceso o pérdida de las capacidades
adquiridas. Se observa una disminución de la velocidad de crecimiento
del cráneo
(de tamaño normal al nacimiento) con respecto al resto del cuerpo entre
los primeros 5 y 48 meses de vida.
Se ha observado un desarrollo psicomotor normal dentro de los
primeros 5 meses de vida. Posteriormente se deterioran las capacidades
manuales anteriormente desarrolladas y aparecen movimientos
estereotipados de las manos (agitarlas, morderlas, retorcerlas). Se observa
también una progresiva pérdida de interés por el entorno social, que en
algunos casos reaparece con la adolescencia.
Otro síntoma, la apraxia -la incapacidad de realizar funciones
motoras- es quizás la característica más debilitante del síndrome de
Rett. La apraxia interfiere con todos los movimientos del cuerpo,
incluyendo la fijación de la mirada y el habla.
Los individuos que padecen del síndrome de Rett a menudo presentan
comportamientos autistas en las primeras etapas. Otros síntomas pueden
incluir caminar con la punta de los pies, problemas del sueño; marcha
con amplia base de sustentación (es decir, con las piernas muy
separadas); rechinar o crujir los dientes y dificultad para masticar;
crecimiento retardado; incapacidades cognoscitivas (del aprendizaje e
intelecto) y dificultades en la respiración al estar despierto que se
manifiestan por hiperventilación, apnea y
aspiración de aire.
También pueden estar presentes anomalías del electrocardiograma, epilepsia, rigidez muscular que puede provocar deformidad y atrofias
musculares, deambulación, escoliosis
y retraso del crecimiento.
Tratamiento
No existe una terapia resolutiva para el síndrome de Rett. Sin
embargo, gran parte de los autores cree que el curso de la enfermedad
puede ser modificado por una variedad de terapias dirigidas a retardar
la progresión de las discapacidades motrices y a mejorar las capacidades
de comunicación. Por ello, el suministro de fármacos
se dirige principalmente a contrarrestar el trastorno motor.
Los fármacos se complementan con terapias dirigidas a conseguir
mejorías tanto en el plano educativo y cognitivo como en el conductual, y
en una mejor gestión de las emociones. También son beneficiosas las
sesiones de fisioterapia que mejoran y mantienen el desarrollo motor
adecuado del niño, así como el mantenimiento funcional y psicomotriz de
los movimientos y posturas, las sesiones de terapia ocupacional se
emplean para disminuir el retraso cognitivo.
Pronóstico
A pesar de las dificultades que acarrean los síntomas, la mayoría de
los individuos que padecen el síndrome de Rett continúan viviendo hasta
la edad adulta. Debido a que el trastorno es poco común, se sabe muy
poco sobre la esperanza de vida. A pesar de que se
estima que hay muchas mujeres de edad mediana (entre 40 y 50 años) que
padecen este trastorno, no se han estudiado suficientes casos para
llevar a cabo cálculos exactos sobre la esperanza de vida más allá de la
edad de 40 años.
La evolución y la gravedad del síndrome de Rett es muy variable.
Algunas niñas presentan un trastorno congénito (antes o durante el nacimiento), mientras que otras pueden
presentar una regresión tardía o síntomas más leves.
Ictiosis arlequín es una enfermedad
de la piel extremadamente rara del grupo de las llamadas genodermatosis. Es la
forma de ictiosis congénita más grave, se hace evidente ya
desde el nacimiento y debe su nombre al aspecto que tienen los recién
nacidos con la enfermedad, que recuerda a un disfraz de arlequín.
La ictiosis tipo Arlequín es una enfermedad genética rara de la piel
caracterizada por escamas grandes y gruesas que aparecen en toda la
piel, como a su vez se nace con los párpados volteados por lo que en
lugar de ojos se observan los párpados totalmente rojos. Se asocia
generalmente deformidades faciales características y a menudo anomalías
en otras partes del cuerpo, especialmente en el tórax.
Se debe a una alteración de la queratinización cuyo mecanismo
fisiopatológico se desconoce, pero se piensa que existe una disgenesia
de la capa lamelar, probablemente debida a anomalías de los lípidos
cutáneos, que da lugar a una hiperqueratosis folicular masiva.
Las complicaciones clínicas más importantes se producen a causa del
fallo de la función de barrera que la piel tiene en condiciones normales
e incluyen sepsis (infección o contaminación generalizada) y
deshidratación que conducen a hipernatremia (aumento anormal de sodio en
sangre) y malnutrición.
Los niños nacen con constricción marcada de tórax y abdomen con las
correspondientes dificultades respiratorias y de alimentación.
El pronóstico es malo, ya que la mayoría fallece a los pocos días o
semanas del nacimiento. Aunque existen casos excepcionales en donde la
persona llega a la adolescencia, pero necesita de tratamientos
especiales.
Es posible el diagnóstico prenatal mediante ecografía, fetoscopia
(procedimiento mediante el cual se puede observar directamente al feto
dentro del útero, mediante el uso de un fetoscopio introducido a través
de una incisión bajo anestesia local) y amniocentesis (procedimiento
obstétrico mediante el cual se extrae una pequeña cantidad de liquido
amniótico para su posterior análisis).
Tratamiento
El tratamiento inicial se centra en atenuar el trastorno de la
función de barrera de la piel, mediante medidas de hidratación,
tratamiento antibiótico ante los signos precoces de infección y medidas
de soporte respiratorio y nutricionales.
Entre las medidas de soporte cutáneas, en el tratamiento de cualquier
tipo de ictiosis, resulta útil reducir al mínimo los baños, emplear,
sólo en los pliegues cutáneos, jabones que no contengan hexaclorofeno
dado que los pacientes presentan mayor capacidad de absorción y, por
tanto, mayor riesgo de toxicidad, aplicar dos veces al día emolientes,
del tipo de la vaselina simple, aceite mineral o lociones conteniendo
urea o propilenglicol; esta aplicación debe hacerse sobre todo después
del baño mientras la piel está todavía húmeda y los productos se deben
dejar retirando el exceso mediante golpecitos con la toalla o mantenerse
como mínimo 10 minutos. Otras sustancias útiles incluyen el gel de
ácido salicílico al 5%, preparados con vaselina hidrofílica y agua a
partes iguales, así como cremas que contengan ácido a-hidroxi láctico,
glicólico y/o pirúvico en diversas bases.
El uso precoz de retinoides tópicos y en los casos muy severos por
vía oral, parece haber mejorado la supervivencia en alrededor de un año,
en los niños que sobreviven la ictiosis parece evolucionar hacia una
eritrodermia ictiosiforme congénita no ampollosa.
A diferencia de las otras ictiosis congénitas el defecto genético no
ha sido identificado. Se hereda como un rasgo genético autosómico
recesivo.
El síndrome de Tourette es un trastorno neuropsiquiátrico heredado con inicio en la infancia, caracterizado por múltiples tics físicos (motores) y vocales (fónicos). Estos tics característicamente aumentan y disminuyen; se pueden suprimir temporalmente, y son precedidos por un impulso premonitorio. El síndrome de Tourette se define como parte de un espectro de trastornos por tics, que incluye tics transitorios y crónicos.
El síndrome de Tourette se consideraba un raro y extraño síndrome, a menudo asociado con la exclamación de palabras obscenas o comentarios socialmente inapropiados y despectivos pero este síntoma está sólo presente en una pequeña minoría de afectados.Entre 0,4% y el 3,8% de los niños de 5 a 18 años pueden tener el síndrome de Tourette. Un Tourette grave en la edad adulta es una rareza, y el síndrome de Tourette no afecta negativamente a la inteligencia o esperanza de vida.
Por lo general, los síntomas del síndrome de Tourette se manifiestan en el individuo antes de los 18 años de edad. Puede afectar a personas de cualquier grupo étnico y de cualquier sexo, aunque los varones lo sufren unas 3 ó 4 veces más que las mujeres.
El curso natural de la enfermedad varía entre pacientes. A pesar de que los síntomas oscilan entre leves hasta muy severos, en la mayoría de los casos son moderados.
Causas
Aunque la causa del síndrome de Tourette es desconocida, las investigaciones actuales revelan la existencia de anormalidades en ciertas regiones del cerebro (incluyendo los ganglios basales, los lóbulos frontales y la corteza cerebral), los circuitos que hacen interconexión entre esas regiones y los neurotransmisores (dopamina, serotonina y norepinefrina) que llevan a cabo la comunicación entre las células nerviosas. Dada la presentación frecuentemente compleja del síndrome de Tourette, la causa del trastorno seguramente es igualmente compleja.
Tratamiento
Por el hecho de que los síntomas no limitan a la mayoría de los pacientes y su desarrollo procede normalmente, la mayoría de las personas con síndrome de Tourette no requieren medicamentos. No obstante, hay medicamentos disponibles para ayudar a los pacientes cuando los síntomas interfieren con las tareas cotidianas. Además, los medicamentos disponibles solamente pueden reducir síntomas específicos.
Algunos pacientes que necesitan medicamentos para reducir la frecuencia e intensidad de los tics, pueden ser tratados con fármacos neurolépticos como haloperidol y pimocida. Se administran estos fármacos usualmente en dosis muy pequeñas que se aumentan lentamente hasta que se logra el mejor equilibrio posible entre los síntomas y los efectos secundarios.
El uso de fármacos neurolépticos a largo plazo pueden causar un trastorno de movimiento involuntario que se llama discinesia tardía. Sin embargo, esta enfermedad usualmente desaparece al dejar de tomar el medicamento. Los efectos secundarios a corto plazo de haloperidol y pimocida incluyen rigidez muscular, babeo, temblores, falta de expresión facial, movimiento lento y desasosiego. Estos efectos secundarios pueden reducirse mediante fármacos usados comúnmente para tratar la enfermedad de Parkinson. Otros efectos secundarios como fatiga, depresión, ansiedad, aumento de peso y dificultad en pensar claramente pueden ser más molestos.
También hay medicamentos disponibles para tratar algunos de los trastornos asociados con el ST. Estimulantes tales como metilfenidato, pemolina y dextroanfetamina, usualmente recetados para el trastorno de déficit de la atención, son algo efectivos, pero su uso es controvertido porque se ha informado que éstos aumentan los tics. Para las conductas obsesivo-compulsivas que significativamente interfieren con el funcionamiento cotidiano se puede recetar fluoxetina, clomipramina, sertralina y paroxetina.
El síndrome de Tourette se puede tratar mediante ejercicios de respiración. Duplicar la frecuencia respiratoria normal del paciente e igualmente duplicar en cantidad cada inhalación. Este tratamiento aumenta las dosis naturales de dopamina, serotonina, norepinefrina y otros neurotransmisores, mejorando también la neurorecepción, así como normalizando el flujo sanguíneo en calidad y cantidad en las zonas del cerebro responsables de los reflejos involuntarios.
Testimonio
Querida extraña, desde hace tiempo estas conmigo, ya no te considero extraña, te conozco, aunque no lo puedo afirmar con seguridad, pues a veces te revelas y haces cosas que me quedo sorprendida.
Al principio todo iba mal, no te quería conmigo, yo misma me discriminaba, era distinta a los demás, cuantas veces habrás oído, "pobrecita de mí", "¿por qué yo?", que ingenua y tonta era ¿verdad? .Si, es cierto que al principio lo pasamos mal, nadie nos comprendía a ninguna de las dos y esto te enfurecía y nos poníamos peor, cuantas luchas y batallas hemos pasado juntas, ¿tú las puedes contar? Yo no, ya he perdido la cuenta. Pero aunque se burlaban de nosotras, en el fondo nos tenían miedo ¿verdad?, porque cuando te enfadabas juntábamos nuestras tuerzas y pobre del que se cruzara por medio.
Ahora que te conozco más permíteme que te describa y si en algo me equivoco, hazme un movimiento, ya que se te da también eso. Se que eres fuerte y orgullosa, pues cuando pasaba algo no querías que te hundieran, eres (en ocasiones) buena compañera, cuando había peleas nos juntábamos como un equipo y juntábamos nuestras fuerzas (que no es poca). A pesar de que todos te conozcan como una persona fuerte, también eres nerviosa y miedica, pues cuando había una pelea, estabas la primera para luchar, pero luego enseguida te ponías a temblar.
Es verdad que en el fondo me has hecho mucho daño, pues por tu culpa he perdido amigas, espera ¿cómo dices? Es verdad, quizás has venido para abrirme los ojos y ver que esas no eran mis amigas, porque ahora, tengo amigas que nos conocen a las dos y nos aceptan. ¡Ay! lo de aceptar, que trabajo cuesta, a mí me costó mucho aceptarte, no te quería conmigo, pero, no me podía enfrentar, eres más fuerte que yo, todavía hay gente que no te aceptan ¿verdad? Pero bueno vamos a darle tiempo, ok? .
Bueno querida amiga no sé el tiempo que estarás conmigo, pero si hemos ganado guerras y batallas, ahora que estás más tranquila no me molestes tanto, aunque, cuando quieres llamar la atención lo haces y bien ¿eh? .Te doy las gracias, quizás te sorprendas, pero sí, porque gracias a ti he conocido a personas maravillosas, que también conviven con amigos/as tuyos. Bueno solo decirte que algún día te irás, y será difícil olvidarte, pero por favor el tiempo que te quedes intenta ser buena y no enfadarte ¿vale? .Sé que lo harás. Gracias querida extraña, gracias.
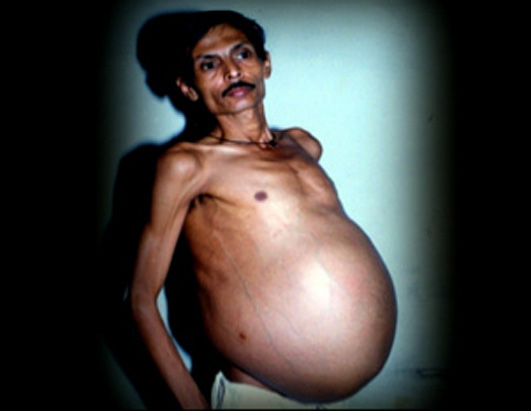
Esta enfermedad se conoce como el síndrome de Fetus in fetus o gemelo parásito y consiste en la formación humanoide creada por un accidente en cierto punto de la formación del cigoto antes de la formación del embrión. Es producida en madres embarazadas de gemelos cuando un feto no sobrevive pero queda alojado en el interior del otro.
Se da en un nacimiento entre más de medio millón de nacimientos. Por lo general, el feto parásito no tiene órganos funcionales internos ni siquiera cerebro, sino que se presenta envuelto en lo que parece una especie de huevo ya que es una cubierta dura, cuando es extraído de ésta tiene un aspecto blanco y muere al poco tiempo ya que es un tejido totalmente dependiente de su hospedador.
Causa un gran riesgo para su hospedador puesto que presenta una carga muerta maciza de hasta un kilogramo que el individuo se ve obligado a cargar y puede causar efectos negativos en su organismo.
Suele diagnosticarse en niños ya que se manifiesta en los primeros años de crecimiento, sin embargo, también hay casos en los que no es notable hasta una edad avanzada. El caso de Fetus in fetus más curioso es en el que el hospedador tenía 47 años.
Síntomas
Generalmente se presenta como
una tumoración no dolorosa en abdomen aunque tambien se han presentado
casos en que se han encontrado en escroto, hígado, riñones, e incluso
dentro de la cavidad craneal. Usualmente es un solo feto el que se
encuentra y puede llegar a pesar unos 1.8 kg y tambien 39 kg.
El primer médico científico en
reportar los limites de diferenciación fue Willis en 1948, que
estableció 2 requisitos importantes para considerar una tumoración
fetiforme como “Fetus in feto”. Y esta son:
1.Debía
haber una columna vertebral que demuestre que el feto ha pasado de un
estudio primario de gastrulación involucrando formación del tubo neural,
con simétrico desarrollo alrededor del axis. 2.Los órganos debían haberse desarrollado de manera sincrónica que permitiera que todos alcanzaran igual grado de maduración.
En la mayoría de los casos de “fetos in fetu”
reportados se han podido demostrar la presencia de una columna
vertebral. Sin embargo, en la mayoría de los casos la maduración de los
órganos no ha sido sincrónica ni todos los órganos están bien
diferenciados.
Estos
hallazgos son atribuidos a fenómenos del feto en la vascularización que
son típicos en el “fetos in fetu”, indicando que la ausencia de una
columna vertebral no demuestra que no halla existido en un estadío más
temprano de maduración previo al nacimiento.
En
la actualidad la mayoría de los autores consideran que “fetos in fetu” y
teratoma no son distintas entidades si no 2 aspectos de la misma
patología en diferentes estados de maduración.
La única solución o tratamiento es la extirpación del feto del interior del huésped el cual, por lo general, puede continuar tras esto con su vida normal.
Casos
Hay muchos casos destacados de esta anomalía. Como hemos dicho suele ser detectada a edades tempranas pero hay veces que no se trata hasta que el individuo no ha alcanzado una edad adulta. Por ejemplo, el caso de:
-Sanju Bhagat nacido en la India y de 36 años de edad. Su abultado
vientre le confería un aspecto de embarazado y en cierto modo era así
pues dentro de él estaba creciendo su gemelo. El feto dentro de él estaba
seriamente malformado y su supervivencia era imposible, a su vez ponía
en peligro la vida de su huesped pues su crecimiento inusual dependía
plenamente de su "hermano" ya formado.
-Otro caso destacable es el de la extirpación de un feto de 700 gramos
del vientre de un niño de tres años. El feto extraído presentaba un
cuerpo con extremidades ''normales'' pero ningún órgano interno,
medía unos 25 cm y pesaba 1,2 kilogramos. El único problema que
presentó su extracción fue que el feto se encontraba unido a un riñón
La diprosopiaes un desorden muy raro en el cual el afectado forma durante su gestación dos caras. Los bebés con este síndrome presentan extremidades normales pero la mayoría de sus rasgos faciales duplicadosen distintas proporciones; pueden tener cuatro orejas y tres ojos o dos bocas separadas y dos narices. En un principio se pensaba que esto se producía por la fusión de dos embriones, pero los estudios sobre esta enfermedad han causado el descarte de esta opción en favor de otras entre las que destaca la que defiende que esta anomalía es causada por una proteína que determina la formación facial del feto y, si ésta existe en cantidades excesivas, puede provocar la formación de dos caras. En su defecto, provocaría la falta de algunos rasgos. Las criaturas que nacen con este defecto fallecen a los pocos días de vida aunque, por suerte, es algo fácilmente detectable en las primeras semanas de formación.
Síntomas
Aunque normalmente se entiende por síntomas a carencias que no son visibles a simple vista, los síntomas de esta enfermedad sí lo son. Entre los más característicos encontramos:
-El labio leporino(condición que se caracteriza por una fisura congénita en el labio superior) -El paladar hendido(fisura congénita en la línea media del paladar) -Anomalías oculares. -Anomalías faciales. -Cambios en la pigmentación de la retina. -Anencefalia (defecto del tubo neural)
Esta enfermedad no tiene tratamiento posible.
Testimonio No se han diagnosticado muchos casos de diprosopia, menos de dos mil en todo el mundo. Aunque habría casos más recientes vamos a conocer el caso de un hombre que sufrió esta enfermedad hace más de un siglo, teniendo en cuenta que en este tiempo no se conocía aún nada de esta enfermedad:
Edward Mordrake se afirma que era un heredero del siglo XIX a uno de los títulos de nobleza en Inglaterra que es conocido por tener un rostro adicional en la parte posterior de la cabeza.
Según la historia, el rostro adicional no podía comer, ni hablar pero
podía llorar y reír. Edward le rogó a los médicos que le quitaran al
gemelo diabólico, según él, porque, supuestamente, le susurraba cosas horribles durante la noche, pero ningún médico se atrevió a intentarlo. A los 23 años, el jóven no aguantó más y se suicidó.
''Una de las historias más raras así como de las más melancólicas de
la deformidad humana es la de Edward Mordrake, quien iba a ser el
heredero de una de las familias más nobles de Inglaterra. Sin embargo
nunca reclamó el título y se suicidó a los veintitrés años. Vivía en un
retiro absoluto, evitando las visitas incluso de los miembros de su
familia. Era un joven de grandes conocimientos, un buen estudiante y un
músico de rara habilidad. Su figura era remarcable por su gracia
natural. Pero en
la parte de atrás de su cabeza había otra cara, la de una chica muy
guapa “adorable como un sueño, atroz como un demonio”. El rostro
femenino era una mera máscara, “ocupando sólo una pequeña zona de la
parte posterior del cráneo, aunque mostrando signos de inteligencia de
aire maligno”. Se la había visto sonriendo y burlándose mientras Mordrake lloraba.Sus
ojos seguían los movimientos del espectador, y sus labios se movían sin
cesar. La voz era inaudible pero Mordrake aseguraba que durante la
noche no podía conciliar el sueño debido a los odiosos susurros de su
“gemela diabólica” como él la llamaba, “que nunca duerme, pero
que me habla de tales cosas de las que sólo se oyen en el infierno''. La
imaginación no puede concebir las tentaciones espantosas en las que me
envuelve. Por alguna imperdonable maldad de mis antepasados estoy cosido
a este demonio – porque estoy seguro que es un demonio. Yo ruego y
suplico para que lo eliminéis del mundo, aunque yo muera”. Estas eran
las palabras del desventurado Mordrake a Manvers y Treadwell, sus
médicos. Aunque lo vigilaban constantemente consiguió procurarse veneno,
debido a lo cual murió, dejando una carta en la que pedía que la “cara
demoníaca” fuera destruida antes de su funeral, “para que no continuase
con sus espantosos susurros en la tumba”. Por petición propia fue
enterrado en tierra baldía, sin ninguna lápida o marca que dejara
constancia de su tumba''.
Edward Mordrake
Ahora dejaremos un vídeo de un caso de diprosopia más reciente:
El síndrome de Klippel-Trenaunay es una enfermedad poco
frecuente, con afección vascular en varios niveles. Un porcentaje bajo de
pacientes presenta hemorragia a través de las várices colónicas que puede
condicionar anemia crónica o hemorragia aguda con compromiso hemodinámico. Esta
complicación amerita tratamiento quirúrgico con resección del segmento
afectado, complementado con alguna medida local para las várices residuales en
el recto.
· Síntomas
Aunque la causa y los procesos que rodean el síndrome de
Klippel-Trenaunay-Weber son poco
conocidos, el defecto congénito que se diagnostica por la presencia de una
combinación de estos síntomas. Una o más manchas distintivas en vino de oporto
con bordes bien definidos
-Venas varicosas
La hipertrofia de los tejidos blandos y óseos, que
pueden llevar a gigantismo locales o disminución.
-Un mal desarrollo del sistema linfático
Estos casos son muy raros y pueden ser clasificados como
síndrome de Klippel-Trenaunay atípico.
Hay que tener en cuenta que SKTW puede afectar los vasos
sanguíneos, los vasos linfáticos, o ambos. La condición más comúnmente se
presenta con una mezcla de los dos. Los que tienen implicaciones venosas están
sujetos a un estilo de vida más difícil debido al aumento del dolor y las
complicaciones.
El defecto de nacimiento afecta a hombres y mujeres por
igual, y no se limita a ningún grupo racial. No se está seguro si es genético
en la naturaleza, aunque las pruebas están en curso.
SKTW también se asocia con taquicardia en las
mujeres afectadas. La embolia pulmonar y trombosis en el 22% de los pacientes
ha sido reportada. Más de 100 mujeres con SKTW han reportado palpitaciones
durante los cambios hormonales, a menudo mal diagnosticados como trastorno de
pánico. SKTW no debe confundirse con el síndrome de Parkes-Weber. La ausencia
de la fístula AV separa sktw del síndrome de Parkes-Weber.
· Tratamiento
SKTW es un síndrome complejo, y no hay un tratamiento que
sea aplicable para todos. El tratamiento se decide sobre una base caso por caso
con los médicos y la persona.
En la actualidad, muchos de los síntomas se pueden tratar,
pero no hay cura para el Síndrome de Klippel-Trenaunay-Weber.
-Quirúrgico
La reducción de volumen ha sido el tratamiento más
utilizado para el síndrome, y se ha utilizado durante décadas. Se ha avanzado
en este método en el transcurso de las dos décadas pasadas, pero sigue siendo
un procedimiento invasivo, y tiene muchas complicaciones. Como en la actualidad
existen otras opciones para los pacientes SKTW, este método se reserva generalmente
como último recurso.
Mayo Clinic ha reportado la mayor experiencia en la
gestión de SKTW con cirugía mayor. En 39 años el equipo de cirugía de la
Clínica Mayo ha evaluado 252 casos consecutivos de SKTW, de los cuales sólo 145
(57,5%) podrían ser tratados con cirugía primaria. La tasa de éxito inmediato
para el tratamiento de venas varicosas era sólo del 40%, la extirpación de la
malformación vascular fue posible en 60%, reducción del volumen de operaciones
en el 65%, y la corrección de la deformidad ósea y la corrección de longitud de
las extremidades (epifisiodesis) tenía 90% de éxito. Todos los procedimientos
han demostrado alta tasa de recurrencia en el seguimiento. Los estudios
demuestran que el manejo quirúrgico primario de Mayo Clinic de SKTW tiene sus
limitaciones y los tratamientos no quirúrgicos deben ser desarrollados con el
fin de ofrecer una mejor calidad de vida de estos pacientes. Cirugía mayor
incluidas las amputaciones y la cirugía de reducción de volumen, no parecen
ofrecer ningún beneficio a largo plazo.
-No Quirúrgico
La escleroterapia es un tratamiento para las venas
y las malformaciones vasculares específicas en el área afectada e implica la
inyección de una sustancia química en las venas anormales que causa el
engrosamiento y la obstrucción de los vasos objetivo. Estos tratamientos pueden
permitir el flujo normal de sangre. Es un procedimiento médico no quirúrgico y
no es tan invasiva como la reducción de volumen. La Escleroterapia guiada por
ultrasonido de espuma es el nuevo tratamiento que podría cerrar muchas malformaciones
vasculares grandes. las operaciones de reducción de volumen pueden dar
lugar a deformidades importantes y tienen un alto potencial de recurrencia y
lesiones nerviosas.
· Testimonio
"Tengo 45 años, y padezco un síndrome de klipel-Trenaunay en
la pierna izquierda desde mi nacimiento. La verdad es que ha sido una lucha
constante, y a fecha de hoy sigo igual, eso sí, pasando por un buen número de
operaciones que aún no sé si sirvieron para algo, aunque me gusta pensar que
sí, un parón largo de unos 20 años en los que ningún médico me daba ni
solución, ni tratamiento. Hasta el nombre y todos sus síntomas lo he sabido
hace 8 años, pues hasta ese momento, lo que yo padecía era un
"angioma" sin más. En 2005 me sometí a una investigación en la
Clínica Universitaria de Navarra y aún sigo llendo alguna vez al año aunque ya
no es investigación, y si antes al ser beca de investigación era gratis, desde
hace 3 años tengo que pagar si quiero que sigan viéndome y no quedar abandonada
de nuevo. Ni qué decir tiene que ese dinero que me piden, tengo que hacer
virguerías para conseguirlo, hasta tal punto que preveo que este año tendré que
dejar de asistir y de nuevo volver a la "nada" de la seguridad
social. Un saludo a todos"